DNA甲基化对细胞周期的调控
摘要 细胞周期(cell cycle)是一种非常复杂和精细的调节过程, 该过程是连续而不可逆的。细胞周期紊乱与很多疾病的发生发展有关, 如肿瘤等。目前的研究发现, 表观遗传学(epigenetics)可以通过影响细胞周期关键性调控因素而调控细胞周期。DNA甲基化(DNA methylation)是最常见的表观遗传修饰方式, 其在基因的转录、基因组的稳定性和细胞周期的调控中发挥重要的作用。因此, 该文重点对近年来DNA甲基化在细胞周期调控中的研究进展作一综述, 以期为提高肿瘤等疾病的临床诊断和治疗水平提供理论依据。
关键词 表观遗传学; DNA甲基化; 细胞周期
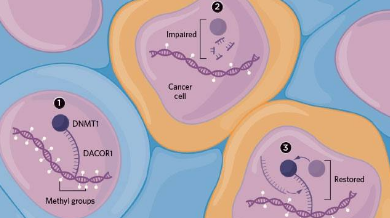
表观遗传学(epigenetics)修饰是指在DNA序列没有发生变化的情况下, 基因功能发生可遗传变化, 并最终导致了表型的变化。表观遗传学修饰的方式主要包括DNA甲基化、组蛋白修饰、非编码RNA、染色质重塑及基因组蛋白印记。DNA甲基化(DNA methylation)是最早发现的表观遗传修饰之一[1], 它在调控基因转录水平、染色体结构的稳定性、基因印记和染色体失活等方面发挥重要的作用[2]。近年来的研究发现, DNA甲基化异常会导致细胞周期紊乱, 从而引起疾病的发生, 如肿瘤等。
1 DNA甲基化
DNA甲基化是哺乳动物基因组主要的表观遗传学调节机制。DNA甲基化是指在DNA甲基转移酶(DNMTs)的催化作用下, 将S-腺苷甲硫氨酸 (SAM)中的甲基转移到胞嘧啶的第5位碳原子上, 形成5-甲基胞嘧啶(5-methylcytosine, 5-mC)[4]。已知哺乳动物体内共有5种DNMTs家族成员: DNMT1、 DNMT2、DNMT3A、DNMT3B和DNMT3L。其中, DNMT1是维持甲基化酶, DNMT3A和DNMT3B是重新甲基化酶。研究发现, DNMT1、DNMT3A和 DNMT3B除了能识别5-mC的生物功能外, 还参与了基因组中5-mC的维持和产生机制[5]。在脊椎动物中, CpG二核苷酸是DNA甲基化发生的主要位点。其中, CpG岛(CpG island)是指基因上富含CpG的序列, 常位于转录调控区附近。
DNA甲基化对基因表达的调节主要通过两种途径完成, 一是通过甲基化的CpG二核苷酸来影响 DNA结构, 进而直接阻碍转录因子与靶基因的结合; 二是甲基化结合蛋白(如MeCP2)与基因中甲基化的 CpG二核苷酸相结合, 诱导染色体的状态改变, 从而抑制基因的转录。DNA甲基化模式是动态的, 以适应环境等的变化[6]。研究表明, 肿瘤等疾病是细胞周期异常性疾病, 异常甲基化影响细胞分裂的生物学稳定性[7]。CpG岛的高甲基化可导致抑癌基因(如参与细胞周期的调控、DNA修复和细胞凋亡等的基因) 沉默; CpG低甲基化可导致癌基因活化[8]。另外, 在肿瘤细胞中DNMTs的活性增高可导致未甲基化的抑癌基因启动子CpG岛的甲基化程度增高[9]。
2 细胞周期及其调控
细胞周期(cell cycle)是指正常连续分裂的细胞从一次有丝分裂结束到下一次有丝分裂结束的过程。根据各个时期的特点不同, 它可以将遗传信息 DNA精确地复制并通过有丝分裂的方式分配到子代细胞中。细胞周期包括静止期(G0期)、DNA合成前期(G1期)、DNA合成期(S期)、DNA合成后期(G2 期)和有丝分裂期(M期)。真核细胞经常受各种不利环境因素(如电离辐射、化学毒物、紫外线等)的影响而导致DNA突变, 从而引起肿瘤及其他疾病的发生。为确保细胞周期的正常运行和遗传信息的稳定遗传, 细胞在长期的进化过程中逐渐形成了一整套细胞周期正常运行的机制, 其中最重要的是细胞周期检测点, 也被称为DNA损伤检测点。数据显示, 研究最多的是G1/S期检测点、G2/M期检查点、中/后期检查点(又称纺锤体组装检查点)。这些检测点使细胞周期进程减慢或阻滞细胞周期进程, 以提供给细胞充足的时间用于修复受损的DNA, 从而保证细胞周期有序运行, 以应对不利因素的影响。然而, 不利的环境等可能引起检测点调控异常, 从而导致肿瘤[10]、帕金森病[11]、阿尔茨海默病[12]等疾病的发生。
3 DNA甲基化对细胞周期检测点的调控
3.1 DNA甲基化对G1/S检测点的调控
细胞周期中G1时程的长短与细胞增殖分化状态密切相关[13]。Gl期检查点是细胞周期调控的关键, Gl期细胞周期蛋白的异常表达很可能引起细胞周期失调, 从而促进疾病的发生。细胞周期的调控是通过有正向调节作用的细胞周期蛋白依赖激酶(cyclin dependent kinases, CDKs)与负向调节作用的细胞周期蛋白依赖性激酶抑制因子(cyclin dependent kinase inhibitions, CDKI)相互作用来实现的。CDK使抑癌基因Rb的蛋白产物磷酸化, 使细胞进入S期; 而CDKI 则抑制CDK活性, 阻断Rb磷酸化, 使细胞停滞于G1 期, 进而发生分化、凋亡或进入静止期。p16、p27、 p21、p57、p53、PENT等基因是细胞生长周期中的负调节因子, 主要参与G1/S检测点的调控。它们与细胞周期的调控、细胞凋亡、DNA修复、细胞分化等重要的生物学功能有关。
3.1.1 p16 p16是一种抑癌基因, 位于人类第9号染色体21号短臂上, 包含2个内含子和3个外显子。 pRb是遗传性视网膜母细胞瘤中发现的抑癌产物, 当CyclinD与相应CDK结合为激酶复合物时可对 pRb磷酸化, 磷酸化的pRb可释放其结合的转录因子 E2F, E2F因子控制并参与细胞增生、生长、分化、凋亡基因的表达, 这些基因的产物促进细胞进入S 期, p16与cyclinD竞争结合CDK4/6使其失活, 从而抑制Rb蛋白磷酸化, 进而阻止细胞周期由G1期进入S 期[14]。研究发现, p16基因的主要失活机制是p16基因启动子区的超甲基化, CpG岛的超甲基化抑制p16 转录, 进而导致其表达产物p16蛋白的缺失[15]。相反, CPG岛低甲基化可使p16基因回复正常的转录[16]。研究表明, p16基因超甲基化使cyclinD依赖性蛋白激酶的活性增高, 导致Rb蛋白异常磷酸化, 从而使细胞绕过正常细胞周期检查点增加细胞增殖和基因组的不稳定性; 同时, p16基因超甲基化可激活cyclinDlCDK4/CDK6-PRb-E2F的环路, 使其无限制反复进行, 使细胞周期由G1期进入S期, 最终导致细胞过度增殖及肿瘤的发生[17-18]。Umemura等[19]的研究表明, 血浆中p16基因启动子甲基化可在早期肺癌中被发现, 提示抑癌基因甲基化是肿瘤发生的早期事件。
3.1.2 p27 p27基因定位于染色体12p12~13.1, 其编码的蛋白质含有198个氨基酸, 由2个外显子和2个内含子组成。研究表明, G1期是其主要作用点, 具有阻止细胞通过G1/S期转化的“关卡”作用。p27对 CDK的抑制作用表现为三方面: 即p27可抑制CDK 的激活过程、p27也可抑制已经激活的cyclin/Cdk的激酶活性、p27还可通过阻止Rb蛋白磷酸化控制细胞周期G1/S转变的进程。DNA甲基调控主要通过 DNMTs, 该酶作用于p27甲基化酶位点Sma I、Hha I、 Ava I。三个酶的作用位点位于DNA的胞嘧啶鸟瞟呤富含区, 该区域与RNA聚合酶II转录基因的启动子有60%的一致性, 甲基化DNA的胞嘧啶干扰了反式作用因子, 使p27的转录产生障碍[20], 抑制了p27蛋白和mRNA的表达, 导致p27基因沉默, 使细胞大量分化和增生。
3.1.3 p21 p21蛋白是CIP家族中的一员, 人类的 p21基因位于6p21.2, 全长约11 Kb, 含有3个外显子和2个内含子。它是位于p53基因下游的细胞周期素依赖性激酶抑制因子。当DNA损伤发生在Gl期时, p21可通过抑制CDK活性来阻止细胞进入S期, 从而抑制DNA复制; 当DNA损伤发生在S期时, p21可通过使DNA聚合酶的相关因子失活而抑制DNA合成, 从而使细胞修复, 保证遗传物质准确地传递给子代, 从而避免了由于DNA损伤的累积而导致的肿瘤发生的可能性[21]。研究表明, DNA甲基化和组蛋白乙酰化可使p21基因失活[22]。其中, 由于在转录起始位点周围p21启动子含有高密度的甲基化CpG二核苷酸聚集[23], 因此易发生甲基化。此外, p21与Cyclin、 CDK和PCNA以四聚体形式存在, 其中p21蛋白在其第144~151氨基酸上存在PCNA结合区域。PCNA是一种分子量为36 kDa的蛋白质, 为DNA聚合酶的辅助蛋白。p21影响PCNA的功能主要依赖于其抑制了PCNA和其他功能蛋白的结合。目前发现至少有两种DNA代谢酶可以与p21竞争PCNA的结合, 其中包括DNMT1。DNMT1可以直接和PCNA结合, 它们的结合会被p21蛋白第141~160位氨基酸残基阻断, 因此, DNA复制过程中p21蛋白可以调控DNA甲基化的水平。所以通常认为, 在哺乳动物细胞中p21对 DNMT1与PCNA结合是负调控[24-25], p21从PCNA复合物中的丢失可引起DNA损伤修复期间甲基化的异常增加[26]。
3.1.4 p57 人类的p57基因定位于11p15.15上, 含有4个外显子和3个内含子, 其中, 2个外显子位于编 码 区 内。p57与p21、p27蛋白同源性在40%以上, p57、p21、p27共享一个N末端区, 以此结合并抑制周期蛋白-CDK复合物的激酶活性。它主要抑制 CCNE-CDK2、CCNE-CDK3、CCNA-CDK2、 CCND-CDK2等。G1期 和S期激酶复合物[27]阻 止 DNA合成和细胞进入S期来抑制细胞增殖。异常的启动子甲基化修饰, 能导致p57基因沉默。研究表明, 与基因沉默相关的不是CpG岛的边缘区域, 而是转录起始位点周围的区域(300~400 bp)密集甲基化[28]。 Kuang和他的同事[29]进行的一项研究发现, 在不同的白血病细胞系中, 有不同的启动子甲基化状态。在 p57基因启动子甲基化的白血病细胞系中, p57过表达导致细胞生长显著停滞和明显的细胞凋亡, p57部分甲基化只导致细胞生长的适度抑制而对细胞凋亡没有影响。这表明, p57的抑癌特性与其细胞的甲基化状态相关。
3.1.5 p53 哺乳动物细胞在受到DNA损伤后, 细胞周期的进程将会停滞于两个关键点, 即G1/S和G2/ M检测点。抑癌基因p53主要在G1检查点上发挥重要作用。p53作为重要的转录因子在DNA损伤、癌基因活化、应激等作用下, 一方面与下游靶基因的 DNA特异性结合来激活靶基因的转录, 另一方面激活的p53通过细胞周期和诱导细胞凋亡等方式防止肿瘤的发生。转录因子p53翻译后要经过一系列复杂的加工与修饰, 最近研究发现, 甲基化修饰是p53 的活性调节方式之一, 对维持p53的活性与稳定性有极其重要的影响[30]。一个特定的、单一的赖氨酸甲基化就可以决定p53的活性, 其中, 转录因子p53羧基末端的K372位点被SET9甲基化可促进p53表达, 导致细胞周期阻滞甚至引起凋亡。有研究表明, p53产生的效应和甲基化位点相关, 如p53羧基末端K370 位点被甲基转移酶Smyd2甲基化后, 会阻遏p53蛋白与DNA的结合, 抑制p53的转录[31]。此外, p53的活性还与该位点甲基化程度密切相关。在DNA复制过程中, DNA甲基转移酶抑制剂通过与DNMTs共价结合, 抑制DNMTs的甲基化作用, 使沉默基因重新表达[32]。由于p53高频率的遗传修饰和功能的双重性, 因此, 成为迄今为止最重要的肿瘤抑制因子。当 DNA损伤时, p53负责细胞周期阻滞。当损伤不能被修复时, p53会激活一系列导致细胞凋亡的事件。研究表明, p53基因在所有类型的白血病中高甲基化, 因此, p53可能是癌症甲基化的潜在靶标[33]。
推荐阅读:医学论文准备技巧有哪些
p53基因对维持细胞的周期起重要作用。一方面, p53基因诱导产生的p21和Gadd45可与PCNA结合, 修复损伤的DNA; 另一方面, p53基因还可以通过与RPA结合而抑制RPA与DNA复制起始点的结合, 在G1后期调节DNA复制起始复合物的合成, 在S期对细胞进行负调控。
3.1.6 PENT 第10号染色体缺失性的磷酸酶及张力蛋白同源(phosphatase and tensin homolog deleted on chromosome ten, PTEN)基因位于人类染色体 10q23.3。PTEN是第一个发现具有双特异磷酸酶活性的抑癌基因, 它通过对3,4,5-三磷酸肌醇(PIP3) 的去磷酸化作用来影响PI3K/Akt信号通道, 激活 Caspase-9及p27等蛋白活性, 抑制CDKs活性, 从而使细胞周期停滞在G1期, 引起肿瘤细胞的凋亡, 从而发挥其抗肿瘤作用[34-35]。高表达的PTEN能通过抑制CDK活性使Rb保持去磷酸化并结合E2F的状态, 从而抑制细胞增殖。PTEN蛋白功能的缺失可导致 PI3K下游信号通路过度激活, 引起丝氨酸/苏氨酸激酶和蛋白激酶B(Akt/PKB)堆积。
《DNA甲基化对细胞周期的调控》
- 张璐团队在《安全与环境
- 陈一团队在《安全与环境
- 赵凤怡团队在《安全与环
- 职称论文刊发主体资格的
- 政法论文浅析工会法主体
- 化学在初中教学中的情感
- 中学教育论文思想政治方
- 法治论文投稿法治型市场
最新优质论文
- 收费的sci怎么样
- 投稿过程中有哪些需要注
- 人工智能视觉识别方向论
- 康复治疗相关论文文献
- 管理工程学报是几种核心
- 教学案例评职称加分吗
- 适合农业废弃物处理论文
- 工程单位评中级职称论文
论文发表问题热点
- 抗震研究的论文发表在什
- 汽车修理技师论文逻辑关
- 辽宁期刊形成具有本地特
- 水质监测员评职称要发表
- 2018年农业论文检索方式有
- 哪里有2018中文核心期刊目
- 消防检测机构怎么申报职
- 2015年河北省教育厅关于教











